

식품의약품안전처(처장 김강립)는 국제적으로 표준화 추세인 ‘의약품 설계기반 품질(QbD)’ 제도를 의약품 허가심사 평가 제도에 도입하는 등 의약품 산업 경쟁력 강화를 위한 규제개선 사항을 반영한 「의약품의 품목허가·신고·심사 규정」을 4월 30일자로 개정·시행한다.
주요 내용은 ▲의약품 설계기반 품질(QbD) 적용 품목의 변경허가 간소화 ▲임상·비임상시험 자료 표준형식(CDISC) 제출 근거 마련 ▲신약 품목허가 상담 신청제도 도입 ▲수입 신약 제출자료 간소화 등이다.
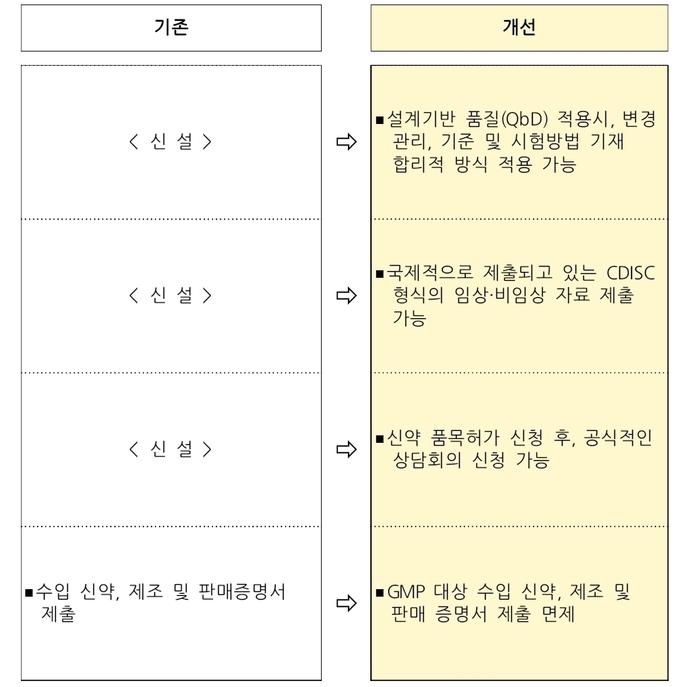
-의약품 설계기반 품질(QbD) 적용 품목의 변경허가 간소화
우수 의약품 개발 및 품질관리를 위해 세계적으로 확산되고 있는 의약품 설계기반 품질(QbD) 제도를 본격 도입한다 해당되는 품목은 제조방법 등 품목허가사항에 QbD 내용을 반영·기재하고 인정 범위 내에서는 유연한 변경관리를 허용하며 완제품 출하 시 일부 시험항목 생략 근거를 제공한다.
의약품 설계기반 품질(QbD) 적용 품목은 기존의 최종 제품 시험을 통한 품질관리 방식이 아니라, 위험평가에 기반해 과학적·통계적으로 설정된 공정별 품질관리 방식을 통해 실시간 출하시험 및 출하가 가능하다.
-임상·비임상시험 자료의 표준형식(CDISC) 제출 근거를 마련
허가심사 시 임상시험과 비임상시험 기초자료 제출을 국제적으로 통용되는 국제표준양식인 CDISC에 따라 제출할 수 있게 된다.
-신약 품목허가 상담 신청 제도 도입
신약 품목허가 신청 후 민원인이 허가·심사 담당 부서에 설명회의 등을 요청할 수 있는 ‘상담 신청제도’를 신설하여 민원 처리의 투명성 및 예측성을 높인다.
-수입 신약 제출자료 간소화
GMP 대상 수입 신약 허가 신청 시 제출해야 했던 수출국 정부 발행 ‘제조 및 판매증명서’ 제출을 면제하여 최초 신약 등에 대해 해외 규제당국의 허가와 해당 국가 내에서의 판매 여부와 별도로 평가할 수 있게 된다.










