
한미약품이 개발한 항암신약 포지오티닙의 글로벌 임상 2상(ZENITH20) 코호트2 연구 결과 및 치료 대안이 없는 응급환자 대상 임상 연구 결과가 유럽종양학회(ESMO) Virtual Congress 2020에서 지난 19일 발표됐다. 한미약품 파트너사 스펙트럼은 코호트2 연구 결과를 바탕으로 FDA에 신약시판허가(NDA)를 위한 미팅 신청을 완료하는 등 포지오티닙의 신속한 허가를 위한 절차를 시작했다. 올해 유럽종양학회는 전세계적 코로나19 여파로 지난 19~21일 온라인으로 진행됐다. 구연으로 발표된 코호트2 연구는 치료 전력이 있는 EGFR/HER2 엑손20 삽입 변이 비소세포폐암 환자 90명을 대상으로 하루 1회 포지오티닙 16mg 경구 투여 방식으로 진행됐다. ITT 분석에서 ORR(객관적반응율)은 27.8%였다. 치료 전력이 있는 환자들의 ORR 최소값 예상치는 17%였으나 실제 ORR 최소값은 18.9%로 유의미한 결과가 확인됐다. DCR(질병조절율)은 70%였으며, 전체 환자의 74%인 67명에서 종양 감소가 확인됐다. 종양 감소 중앙값은 22%였다. 평가 가능한 환자 74명에서 ORR은 35.1%, DCR은 82.4%로 확인됐다. mDOR(
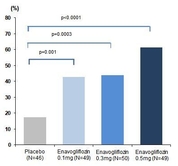
대웅제약(대표 전승호)이 18일부터 19일까지 진행된 ‘2020 ICDM(2020 International Congress of Diabetes and Metabolism)’에서 현재 개발중인 SGLT-2 계열 당뇨병 치료제 ‘이나보글리플로진’의 임상 2상 결과를 최초로 발표했다. 이번 ICDM(국제 당뇨병 및 대사질환 학술대회)은 대한당뇨병학회 주최, 10주년을 맞은 국제학술대회로 신종 코로나바이러스 감염증(코로나19) 확산방지 차원으로 온라인을 통해 진행됐다. 이나보글리플로진은 혈당이 적절히 조절되지 않는 2형 당뇨병 환자를 대상으로 12주 복용했을 때, 투여 4주차부터 위약대비 당화혈색소(HbA1c) 변화량이 통계적으로 유의한 감소를 보였고, 12주 째에는 위약 대비 약 0.9% 감소했다. 이는 통계적으로 유의한 결과로 기존 서양인들을 대상으로 진행됐던 타사 SGLT2 억제제보다 약 0.2~0.3% 정도의 추가적인 당화혈색소 감소이며 추가 연구가 기대되는 결과다. 대상자별 치료효과를 평가한 12주 때는 당화혈색소가 7.0% 이하로 도달한 환자 비율이 최대 61%에 달해 기존 SGLT-2 억제제보다 효과를 보인 환자 비율이 20% 이상 증가한 결과를 보였
㈜헬릭스미스는 미국식품의약국(FDA)에 유전자치료제 ‘엔젠시스(VM202)’의 당뇨병성 신경병증(DPN) 임상 3-3상 프로토콜을 제출했다고 16일 밝혔다. 이번 DPN 임상 3-3상은 엔젠시스(VM202)의 유효성을 확인하기 위한 임상시험으로서, 복수의 3상 임상 결과를 권고하는 미국 FDA 가이드라인에 따라 기획되었다. 그 내용은 지난 3-2상과 거의 같은데, 다만 장기간(1년) 통증 효과와 안전성 데이터를 수집하기 위해 추적 관찰기간을 1년으로 설정했다. 주평가 지표는 첫 주사 후 6개월째에 통증일기(pain diary)로 측정된 지난 1주일 간의 평균 통증의 감소 효과를 측정한다. 부평가 지표는 (1) 6, 9, 12개월째에 지난 1주일 간의 평균 통증의 감소 효과가 50% 이상인 환자의 비율, (2) 첫 주사 후 9, 12개월째에 통증일기로 측정된 지난 1주일 간의 평균 통증의 감소 효과, (3) 6, 9, 12개월째에 가장 심한 통증의 감소 효과 등이다. 대상 환자는 3-2상과 마찬가지로 프리가발린, 가바펜틴 등 가바펜티노이드 계열 약물을 복용하지 않는 DPN 환자다. 환자 규모는 처음에 152명으로 시작해 50%의 피험자에 대한 자료를 수집한 뒤

사노피 젠자임의 ‘듀피젠트®(성분명: 두필루맙, 유전자재조합)’가 만 12세 이상 청소년 및 성인 중등도-중증 천식 환자를 대상으로 한 3년(96주) 장기 임상연구 데이터를 통해 일관된 치료효과와 안전성 프로파일을 확인했다. 해당 데이터는 2020 유럽호흡기학회 연례학술대회(ERS, European Respiratory Society)의 라이브 세션에서 발표되었다. 미국 국립유대의료센터 천식연구원(National Jewish Cohen Family Asthma Institute) 소속 마이클 웩슬러(Michael Wechsler)박사는 “이번 연구를 통해 듀피젠트®가 중등도-중증 천식 환자들이 경험하는 점진적인 폐 기능 감소를 늦추고 최대 3년 동안 지속적으로 폐 기능을 개선하는 것을 확인했다”며, “듀피젠트® 투여군은 천식 증상을 조절에 성공했고, 응급실 내원을 유발할 수 있는 천식 악화 빈도 또한 감소했다”고 설명했다. 특히 “듀피젠트®는 천식 환자, 그 중에서도 제2형 염증성 천식 환자에서의 호흡 능력을 개선시켰다”며, “이는 장기간의 천식 치료에 있어 듀피젠트®의 치료 효과를 확인한 의미 있는 결과”라고 강조했다. 이번 ERS에서 발표될 연구에는 듀피
㈜헬릭스미스가 유전자치료제 ‘엔젠시스(VM202)’의 근위축성 측삭경화증(ALS, 루게릭병) 미국 임상 2상에 대한 임상시험 수탁기관(CRO)으로 Worldwide Clinical Trials(월드와이드)를 선정했다. 헬릭스미스는 최근 월드와이드와 포괄적 협력계약(Master Service Agreement, MSA)을 체결했다. 이번 CRO 계약 체결에 따라 월드와이드는 미국에서 실시할 엔젠시스(VM202)의 ALS 임상 2상을 관리할 예정이다. 월드와이드는 미국에 본사를 두고 60여개 국가에 네트워크를 보유하고 있는 글로벌 기업이다. 중추신경계, 심혈관 및 대사장애, 일반 의학, 종양학 및 희귀질환에 중점을 두고 미충족 의료수요가 큰 질환을 전문으로 한다. 30년 이상의 중추신경계 연구 지원 경험을 보유한 월드와이드는 ALS 임상시험 관리 분야에서 국제적으로 인정받는 전문 CRO다. 대부분의 ALS는 원인을 알 수 없고 질병의 전개를 점수를 매겨 판단하기 때문에 경험이 많은 전문기관이 아니면 임상시험을 관리하기 매우 어렵다. 근위축성 측삭경화증(ALS)은 치명적인 진행성 신경근육 질환으로 루게릭병이라고도 불린다. 근육의 움직임을 조절하는 운동 뉴런(신경세

㈜지엔티파마(대표 곽병주)가 뇌세포 보호 신약 ‘넬로넴다즈’의 임상 3상에 돌입한다. 신약개발 벤처기업 지엔티파마는 중국 파트너인 헹디안 그룹 아펠로아가 뇌세포 보호 신약 ‘넬로넴다즈’의 임상 3상 시험을 중등도 및 중증 급성 뇌졸중 환자를 대상으로 중국 40개 대학병원에서 개시한다고 10일 밝혔다. 넬로넴다즈 임상 3상 시험은 위약(플라시보) 또는 넬로넴다즈를 1:1의 비율에 따라 이중 맹검방식으로 투약하여 위약 대비 넬로넴다즈의 약효와 안전성을 검증하는 연구다. 발병 후 8 시간 이내의 급성 허혈성 뇌졸중 환자 948명을 대상으로 진행한다. 시험에 참여하는 뇌졸중 환자는 CT 영상으로 허혈성 뇌졸중이 확인되고 미국 국립보건원 뇌졸중 등급(NIHSS)에 따라 9-22 사이의 중등도 또는 중증 장애가 있어야 한다. 시험자는 약물을 투여하기 전에 허혈성 뇌졸중의 유일한 치료법인 혈전용해제tPA(tissue plasminogen activators)나 유로키나제를 투여받는다. 이번 임상 3상 시험은 선행 임상 연구에서 확인된 약효와 안전성에 근거하여, 넬로넴다즈 총 6000 mg을 5일에 걸쳐 투여한 후 90±7일까지 뇌졸중 환자의 장애, 사망, 출혈 등을 정량
한독(대표이사 김영진, 백진기)이 진행하고 있는 고혈압 복합제 임상시험이 2상에 본격 돌입했다. 한독은 사노피와 협력,ARB 계열 이르베사르탄과 CCB 계열 암로디핀 성분의 고혈압 복합제를 개발하고 있다. 현재까지 여러 종류의 ARB-CCB 복합제가 나와 있지만, 이르베사르탄-암로디핀 조합 복합제는 한 품목도 없다. 한독은 올해 3월 식품의약품안전처로부터 임상 2상 계획(IND)을 승인 받았으며 5월 첫 환자 투여를 시작했다. 현재까지 연세대학교 세브란스병원, 서울대학교병원, 고려대학교 안암병원, 가톨릭대학교, 서울성모병원 등 주요 병원을 포함해 약 30개 기관에서 임상연구심의위원회(IRB) 승인을 획득하고 참여자를 모집하고 있다. 이번 임상의 목적은 본태성 고혈압 환자에서 이르베사르탄과 암로디핀의 병용요법과 각각의 단일요법의 유효성과 안전성을 비교 평가하는 것이다. 임상은 만 19세 이상 75세 이하 성인을 대상으로 하며 2021년 2월 완료를 목표로 하고 있다.
사노피와 GSK가 공동 개발 중인 면역증강제 기술 기반 신종 코로나 바이러스 19(이하 코로나19) 백신 후보물질의 1/2상 임상시험을 시작했다고 3일 밝혔다. 이번 백신 후보물질은 사노피의 계절 독감 백신 중 하나와 동일한 재조합 단백질 기반 기술과 GSK의 검증된 팬데믹 면역증강제 기술(pandemic adjuvant technology)을 기반으로 개발되었다. 이번 1/2상 임상시험은 백신 후보물질의 안전성과 내약성, 면역원성(면역반응)을 평가하기 위한 무작위 배정, 이중맹검, 위약 대조 시험이다. 미국 내 11개 연구기관에서 모집된 건강한 성인 440명을 대상으로 진행된다. 양사는 올해 12월 초 임상시험의 첫 결과가 나올 것으로 예상하고 있으며, 이는 2020년 12월의 3상 임상시험 시작을 뒷받침할 것이다. 또한, 코로나19 백신 승인 신청에 필요한 충분한 데이터가 도출될 경우, 2021년 상반기 내 시판 승인을 신청할 예정이다.

대웅제약(대표 전승호)은 신종 코로나바이러스 감염증(코로나19) 치료제로 개발중인 'DWRX2003(성분명 니클로사마이드)의 인도 임상 1상 승인에 이어 3일 현지 건강인 대상 투약을 시작했다고 밝혔다. 첫 투약 그룹에서 현재까지 안전성을 확인했고, 임상시험이 순항 중이다. 이번에 개시한 임상 1상은 현지에서 건강한 피험자 약 30여명을 대상으로 안전성과 내약성을 확인할 예정이다. 특히, 인도에서 확보되는 데이터는 코카시안 대상의 데이터로, 미국과 유럽 등 글로벌 임상시험 진입 시 중요하게 활용될 수 있는 인종간 안전성 및 약물동력학 데이터로 사용될 예정이다. 인도는 미국에 이어 세계에서 두 번째로 코로나19 누적 확진자가 많은 나라이다. 7일 기준으로 하루 확진자가 9만명 대로 올라서는 등 폭증세를 이어가고 있는데, 국가적 방역 통제는 해제된 상황이어서 치료제 개발이 시급한 국가 중 하나이다. 대웅제약은 니클로사마이드 임상 가속화와 신속한 현지 공급을 위해 인도 3위 제약사 맨카인드파마와 라이선스 및 공동개발 협약을 체결한 바 있다. 또한 인도에서의 건강인 대상 안전성 확인과 병렬로, 필리핀에서는 코로나19 감염 환자를 대상으로 한 임상 1상을 승인받아 9

대웅제약이 'DWRX2003(성분명 니클로사마이드)'의 연이은 해외 임상 승인을 통해 글로벌 코로나19 치료제 개발을 본격화하고 있다. 대웅제약(대표 전승호)은 4일(현지시간) 필리핀 식품의약품안전청(PFDA)으로부터 신종 코로나 바이러스 감염증(코로나19) 치료제 'DWRX2003(성분명 니클로사마이드)'의 임상 1상 시험을 승인받았다. 지난 인도 임상 1상 승인에 이은 두 번째 ‘니클로사마이드’ 해외 임상 승인이다. 이번 필리핀 임상 1상은 코로나19 환자 40명을 대상으로 9월 중 첫 투여를 시작한다. ‘DWRX2003’의 안전성, 내약성과 유효성 등 약물 유효성 초기 지표를 확인하는 것에 중점을 둘 예정이다. 필리핀은 동남아시아에서 코로나19 환자 발생이 많은 국가 중 하나이다. 대웅제약은 현지 법인의 허가개발 역량을 활용해 4개월 이상 걸리는 임상 시험 심사 기간을 2개월로 단축해 이례적으로 빠른 승인을 받았다. 이번 임상 1상을 완료 후 2상 임상 시험을 진행할 예정으로 결과 확보 후에는 현지 긴급승인을 신속히 추진하는 한편, 3상 및 허가를 신청할 계획이다. 'DWRX2003(성분명 니클로사마이드)'는 세포의 자가포식 작용을 활성화해 바이러스 증
(주) 메디팜헬스뉴스/등록번호 서울 아 015222/등록일자 2011년 2울 23일/제호 메디팜헬스/발행인 겸 편집인 노재영/발행소 서울특별시 송파구 송파대로 42길 45 메디팜헬스빌딩 1층
발행일자 2011년 3월 3일/청소년보호 책임자 노재영/Tel. 02-701-0019 / Fax. 02-701-0350 /기사접수 imph7777@naver.com
메디팜헬스뉴스의 모든 기사는 저작권의 보호를 받습니다. 따라서 무단사용하는 경우 법에 저촉됩니다.
UPDATE: 2026년 04월 21일 13시 05분








